Science
DNA Replication
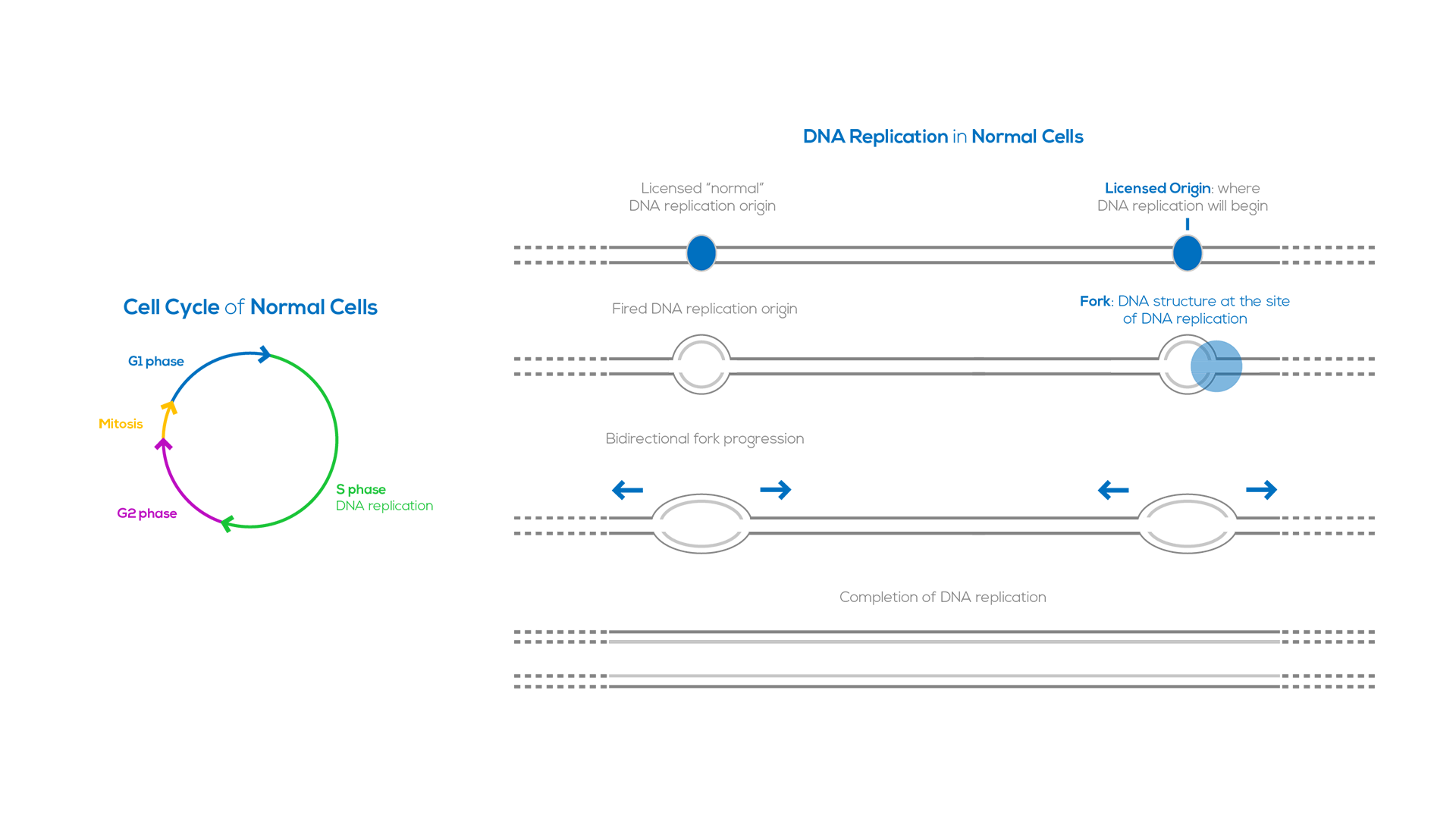
DNA replication is a well-regulated process that duplicates the genome before each cell division. This process begins at specific genomic loci termed “origins of replication’’. DNA replication involves first licensing and then firing of the DNA replication origins. In human cells licensing occurs during the G1 phase of the cell cycle, at which time thousands of replication origins are established along the genome. DNA synthesis, also referred to as origin firing, occurs in S phase and requires activation of specific cyclin-dependent kinases. DNA replication is carried out in a bidirectional fashion; two replication forks start from each origin and proceed in opposite directions. DNA replication is completed when forks from adjacent origins converge.
Oncogene-induced DNA Replication Stress
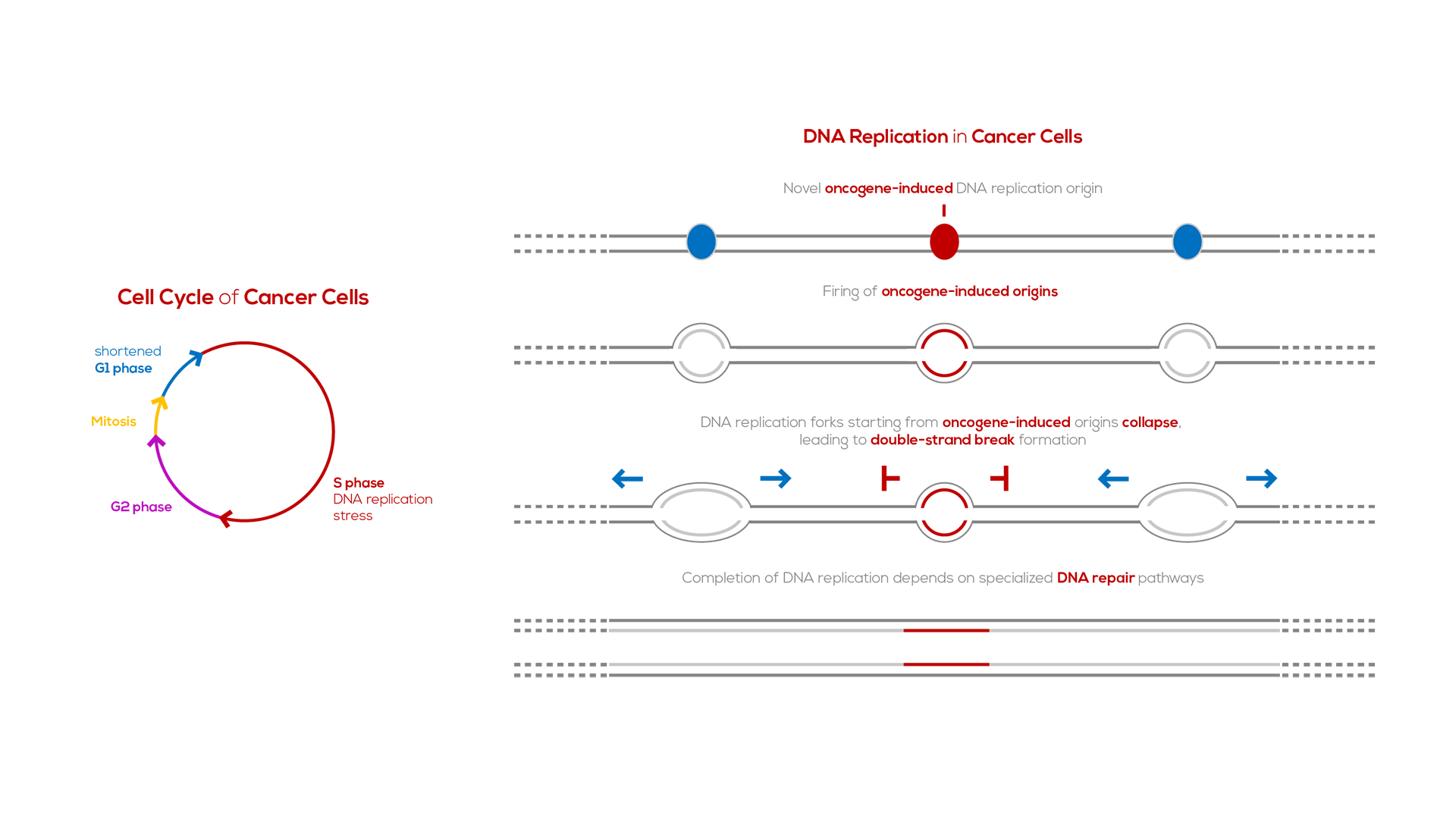
A key step in the development of most cancers is the acquisition of mutations in genes (oncogenes and tumor-suppressor genes) that regulate cellular proliferation. In parallel, in most cancers there are, paradoxically, difficulties in duplicating the genome, due to stalling and collapse of the DNA replication forks, a phenomenon known as “DNA replication stress”. Oncogene activation is one of the main sources of DNA replication stress, which, in turn, leads to DNA breaks and genomic instability1,2. The latter helps drive cancer progression and resistance to therapy, as it allows for the establishment of mutations that favor tumor growth and adaptation to diverse conditions3,4.
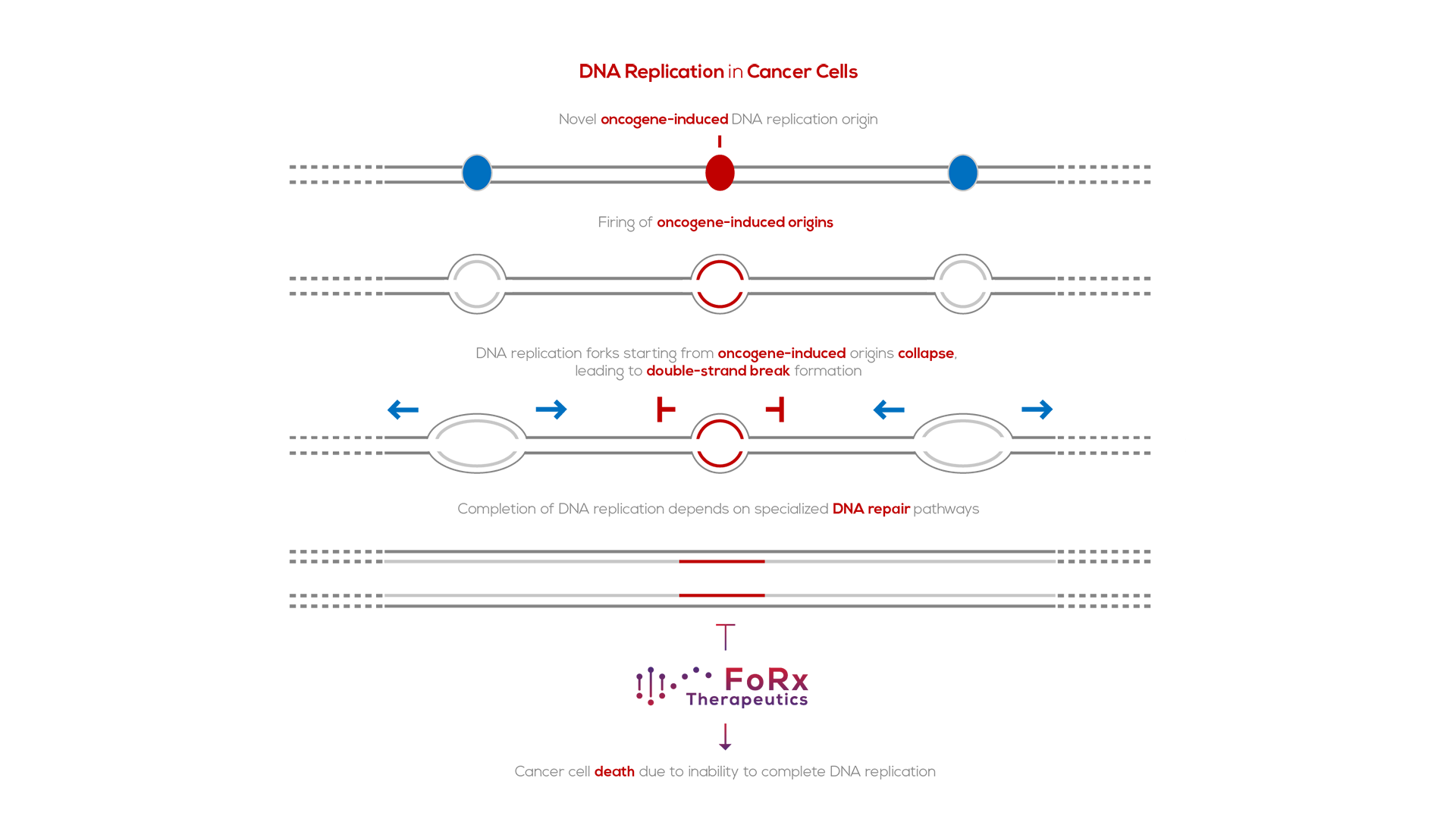
Oncogenes induce DNA replication stress by perturbing the cell cycle. Specifically, oncogenes stimulate premature entry of cells into S phase, the DNA synthesis phase of the cell cycle. As a consequence, specific DNA replication origins, which are silenced in normal cells, fail to be silenced in cancer cells5. These “oncogene-induced DNA replication origins’’ are present in highly-transcribed genes; as such, forks from these origins are prone to collapse due to replication-transcription collisions.
Collapsed DNA replication forks need to be repaired for cells to duplicate their genome. The principal repair pathway for collapsed DNA replication forks is “break-induced replication”6. Since fork collapse is much more frequent in cancer cells than in normal cells, cancer cells are more dependent on break-induced replication for their survival than normal cells.
Synthetic Lethality: Turning Replication Stress against Cancer Cells
Synthetic lethality refers to the genetic principle in which the combination of two genetic perturbations is lethal, where each individually is not. Oncogene-induced DNA replication stress, a hallmark of cancer7, makes cancer cells dependent on repair pathways for collapsed DNA replication forks. FoRx Therapeutics is developing small chemical inhibitors of these repair pathways to specifically target cancer cells, while sparing normal cells.
References
1. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. (2006) Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444: 633-637
2. Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434: 907-913
3. Halazonetis TD, Gorgoulis VG, Bartek J. (2008) An oncogene-induced DNA damage model for cancer development. Science 319: 1352-1355
4. Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montana MF, et al. (2011) Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 18: 1331-1335
5. Macheret M, Halazonetis TD. (2018) Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555: 112-116
6. Sotiriou SK, Kamileri I, Lugli N, Evangelou K, Da-Re C, Huber F, et al. (2016) Mammalian RAD52 functions in break-induced replication repair of collapsed DNA replication forks. Mol. Cell. 64: 1127-1134
7. Macheret M, Halazonetis TD. (2015) DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 10: 425-448